 杭州浙大迪迅生物基因工程有限公司
杭州浙大迪迅生物基因工程有限公司Release date:2021-05-28
JACI
[IF:13.1]
Non–IgE-mediated anaphylaxishttps://doi.org/10.1016/j.jaci.2021.02.012
Abstract:
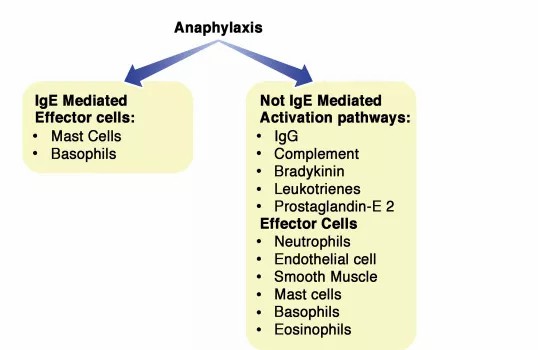
Anaphylaxis is defined as a severe, life-threatening, rapidly evolving, generalized or systemic hypersensitivity reaction.Typical clinical signs include bronchoconstriction, vasodilatation, and hypovolemia, which can lead to respiratory insufficiency, shock, and death.3 The term allergic anaphylaxis is used when the reaction is mediated by an immunologic mechanism (IgE, IgG, and immune complex [IC] complement-related). Based on the involvement of IgE is the reaction then subclassified into IgE-mediated and not IgE-mediated reaction.
Typical clinical signs include bronchoconstriction, vasodilatation, and hypovolemia (alone or in combination), which can lead to respiratory insufficiency, shock, and death. The most prominent cause of anaphylaxis in children is food, whereas in adults it is drugs, especially antibiotics, neuromuscular-blocking agents (NMBAs), nonsteroidal anti-inflammatory drugs (NSAIDs), contrast medium, and biologics.
The classical example of an anaphylactic reaction mediated by IgE antibodies is peanut-induced food anaphylaxis, and it may be referred to as IgE-mediated allergic anaphylaxis. Such anaphylaxis is classically considered to be due to the production of IgE antibodies from B cells after T cells have recognized the antigen and initiated a TH2-type response.
The IgE-mediated pathway is initiated by an allergen that, when bound to specific IgE antibodies, generates a cross-linking of the high-affinity IgE receptor (FcεRI) on mast cells (MCs) and basophils and initiates a signaling cascade that produces the release of mediators, including histamine, as well as preformed cytokines and proteases, and the synthesis and secretion of additional cytokines as well as lipid mediators, such as platelet-activating factor (PAF), leukotrienes, and prostaglandins. The classical anaphylaxis pathway is an example of adaptive immunity. The clinical diagnosis of anaphylaxis to an allergen is based on this IgE paradigm. Higher levels of IgE to a specific allergen are associated with more severe/persistent disease and treatment failure when using immunomodulatory treatments such as immunotherapy or anti-IgE.
However, a considerable percentage of patients who experience anaphylaxis do not present with any evidence of IgE-dependent immune activation. In these patients, there are no signs of IgE-mediated activation of MCs or basophils as suggested by a positive skin test result, presence of allergen-specific IgE, or elevated histamine. In some patients, there is no evidence of MC activation at all; for example, they have normal tryptase. All together these data suggest the existence of other potential mechanisms.
Indeed, in the absence of IgE, anaphylaxis can still develop in mouse models.
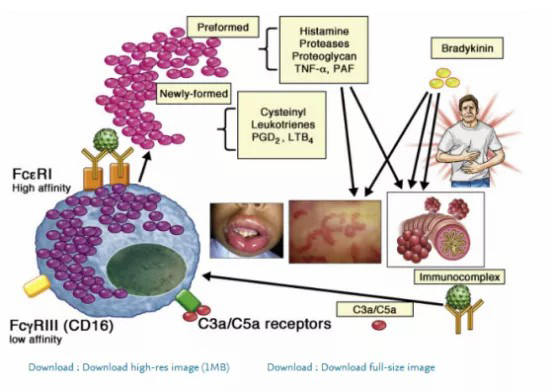
IgE-independent MC/basophil, as well as non-MC/basophil, pathways have been described in mouse models (Fig 1).
MCs are generally considered the most important cell type in driving anaphylactic reactions. They are classified into 2 types on the basis of expression of different proteases in their secretory granules.14 In humans, tryptase and chymase-expressing MCs are found in connective tissue (ie, the skin) and contain various proteases such as tryptase, chymase, carboxypeptidase, and cathepsin. In contrast, tryptase-expressing MCs are found in the lung and gut and express only tryptase.14 All MCs can be activated through the cross-linking of high-affinity IgE receptors (FcεRI); however, even if this appears to be the major contributor to the signs and symptoms associated with hypersensitive and allergic diseases, it likely does not play a major role in the physiological activation of MCs, especially tryptase and chymase-expressing MCs (also known as MCTC), during endogenous stimuli activation to promote vascular hemostasis, pain, itch, and host defense.
In the latter mechanism, tryptase and chymase-expressing MCs respond to complement component fragments of the complement (complement fragment 3a [C3a] and complement fragment 5a [C5a]) and 48/80 (a compound that can mimic in vitro complement activity on MCs), but the endogenous and/or natural exogenous ligands remain unknown (Fig 2). Other mechanisms involve activation through numerous G protein–coupled receptors (GPCRs), the largest group of membrane receptor proteins, and the most common targets of drug therapy and MCs express many of them on their surfaces.
Among these receptors, the one of particular interest is Mas-related G protein–coupled receptor X2 (MRGPRX2, formerly known as MrgX2), which is expressed selectively in MCs in their plasma membrane and intracellular sites and is activated by antimicrobial host defense peptides, neuropeptides, major basic proteins, eosinophil peroxidase, and many US Food and Drug Administration–approved peptidergic drugs.14
The existence of such alternative mechanism to cause anaphylaxis in humans is not definitive, even if some clinical observation may point toward that direction.
For example, rare clinical observations in humans suggest that anaphylaxis arises not only following the IgE/mast cell/basophil axis but also from neutrophils, platelets, endothelial cells activated via various mechanisms such as complement activation, neuropeptide release, IC formation, cytotoxicity, and IgG-dependent reactions, and the involvement of purinergic metabolism with mechanisms not yet fully understood.
Similarly, the effectiveness of anti-IgE in non–IgE-mediated diseases may indicate that IgE may be implicated beyond the classical IgE/allergen mechanism.15 These alternative pathways, as well as those molecules that activate MCs and basophils in a non–IgE-mediated classic fashion, may contribute to the intensity of anaphylactic reactions and may help us to understand why the severity of the anaphylaxis varies widely, despite similar degrees of activation of the IgE pathway.
In this article, we will review pathways that may lead to anaphylaxis in the absence of specific IgE/allergen activation, as well as those independent of MCs and basophils altogether.
Author:
Antonella Cianferoni
2021-5-11 Review
Hangzhou Zheda Dixun Biological Gene Engineering Co., Ltd. Copyright HZKC Technical support 浙ICP備14005341號(hào) (浙)-非經(jīng)營(yíng)性-2019-0050
Address: Rm.201-209, Bldg.2, No.568 Binkang Rd., Binjiang Dist. Tel: 0571-87968248-805 Website:baochunhua.cn
COPYRIGHT©2003-2024 baochunhua.cn CORPORATION. ALL RIGHTS RESERVED


Official WeChat